
General
The batchtool was created out of the needs to apply common neuroimaging pipelines (like SPM, FSL, Freesurfer) on medium sized projects (10-1000 subjects) in a convenient and efficient manner. One of the major objective of NORA's batchtool is to deal with heterogenous data. Typically files (a.k.a. image series) are selected by file patterns, which can iteratively generalized. Errors can be easily tracked by a simple error logging system. For example, it is simple to select a errornous subgroup and rerun a modified job for them, which was corrected for the error. Processing pipelines (batches) are a simple linear series of jobs. Depending on the relationship between individual jobs, they can run serially or in parallel.
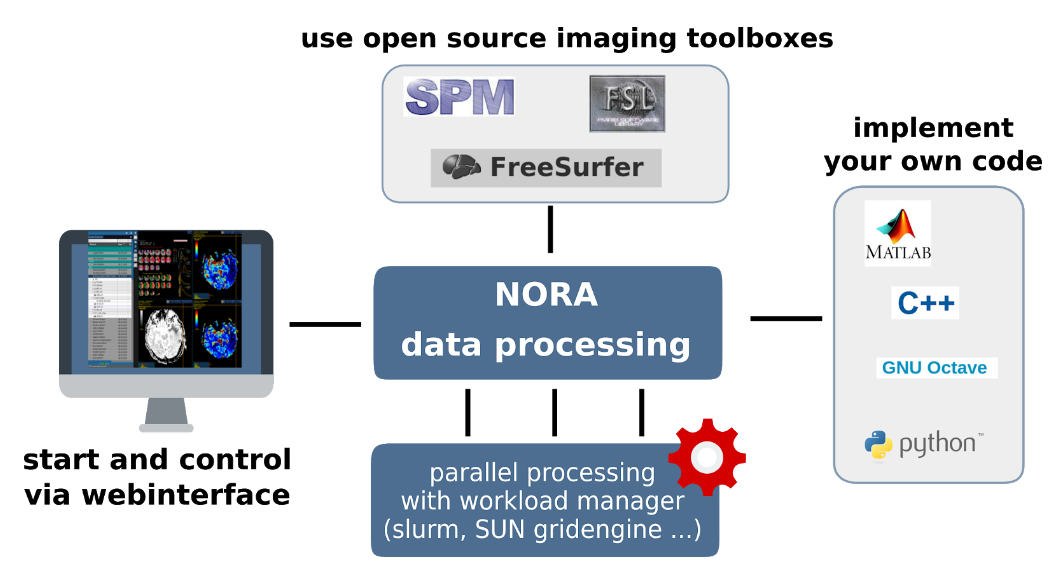
In conclusion, what it provides:
- Convenient selection of subject/study sets to apply certain processing pipelines
- Definitions of inputs via Tags or filepatterns with wildcards
- Composition of processing pipelines based on predefined scripts/jobs (mostly MATLAB), or custom MATLAB/BASH/Python code
- Submission of jobs to a cluster (Slurm/SGE) with direct access to logs and errors
The Batchtool Window
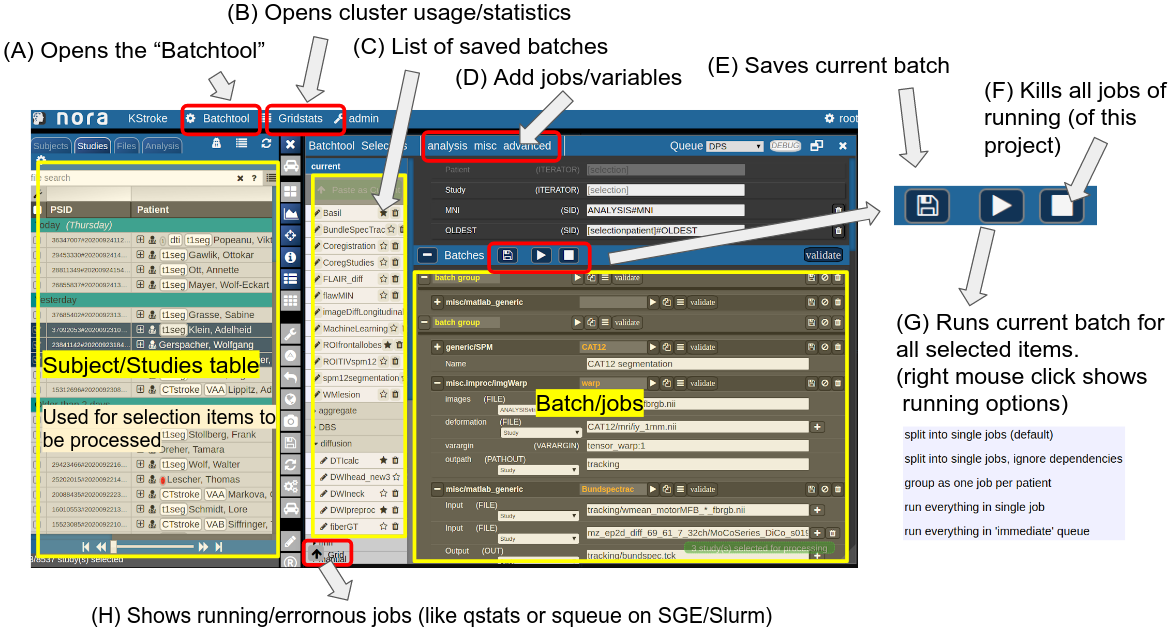
Consider Figure 2 below: the subject/studies table on the left is used for the selection of subject/studies on which you want to run your batch. You can use the filter bars to create the subgroup you want to work on (see Subject/Studies table). Select on "Batchtool" on the top toolbar (A) to open the batchtool. Below you see the structure of the Bacthtool window. It allows to compose the batch out of single jobs. Jobs are added from the menu (D). You can save batches (E), which then appear in the batch list (C). To open an overview of currently running jobs open the "Gridstats" window (B) or (H).
Depending on the selection level (Subjects or Studies) launched jobs are iterated over subjects (patients) or studies. 
In a scenario where you have multiple studies per patient, which have to be linked in some sense, the subject level is appropriate. For example, think of a neuroimaging analysis where you a have a CT study (which contains, e.g. electrode information) and a MR study (which contains soft tissue anatomical information), or, simple a longitudinal analysis. Otherwise, when you have studies, where all studies are treated equally, the study level is appropriate.
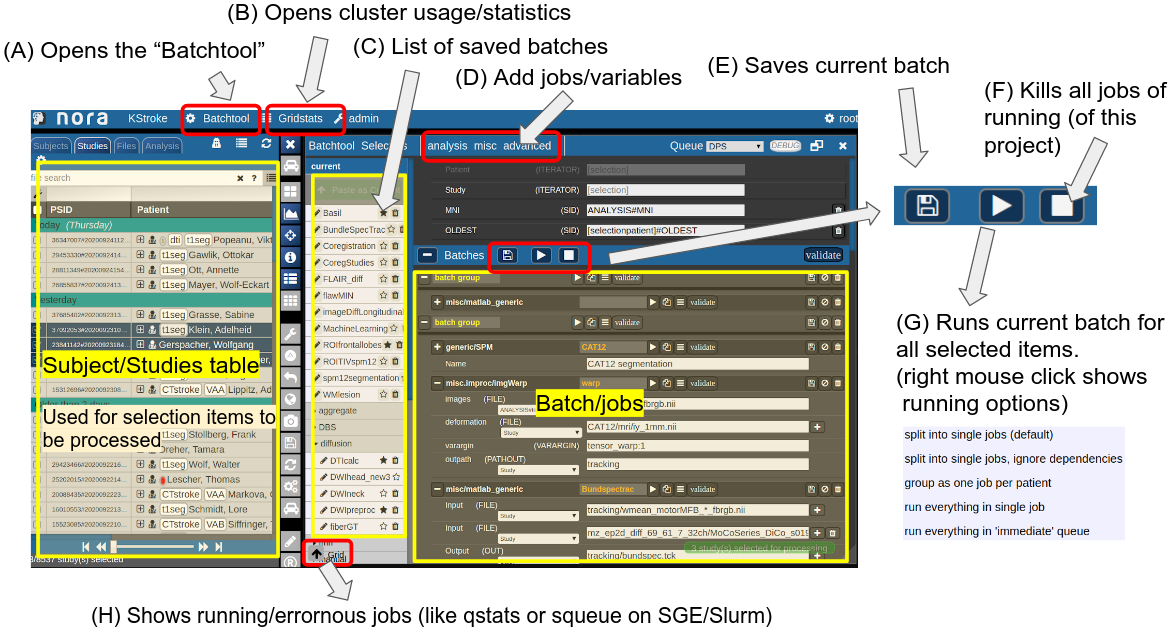 Figure 2: Batchtool overview.
Figure 2: Batchtool overview.
The Anatomy of a Job
A job consists of a list of arguments. There are several types of arguments:
-
FILE
All input images/series (or any other type of files) are given as FILE arguments. Usually you give aFILEfile pattern instead of an explicit filename. AFIlefIle pattern is a combination of subfolders, filename and wildcards. For example:subfolder1/subf*t1*/s0__.nii.ItIcanreferscontainsto all files contained in a folder starting with t1 and whose filename matches "s0___.nii" . The asterisks (*)asis a placeholder for an arbitrary character sequence,or aan underscore "_" for a singlecharacter placeholder.character. Internally, the wildcards are the same as for SQL "like" statement (the '*' is replaced by '%'). A FILE argument also includes a reference to a study or patient. Depending on the selection level (subject or study), different "study references" are possible. -
OUT
A name of a file including the subfolder. No wildcards are allowed here. -
PATHOUT
Same as OUT but refers to foldername instead of a filename. - NUMERIC
- STRING
- LOGICAL
- OPTION
Study References and study selectors
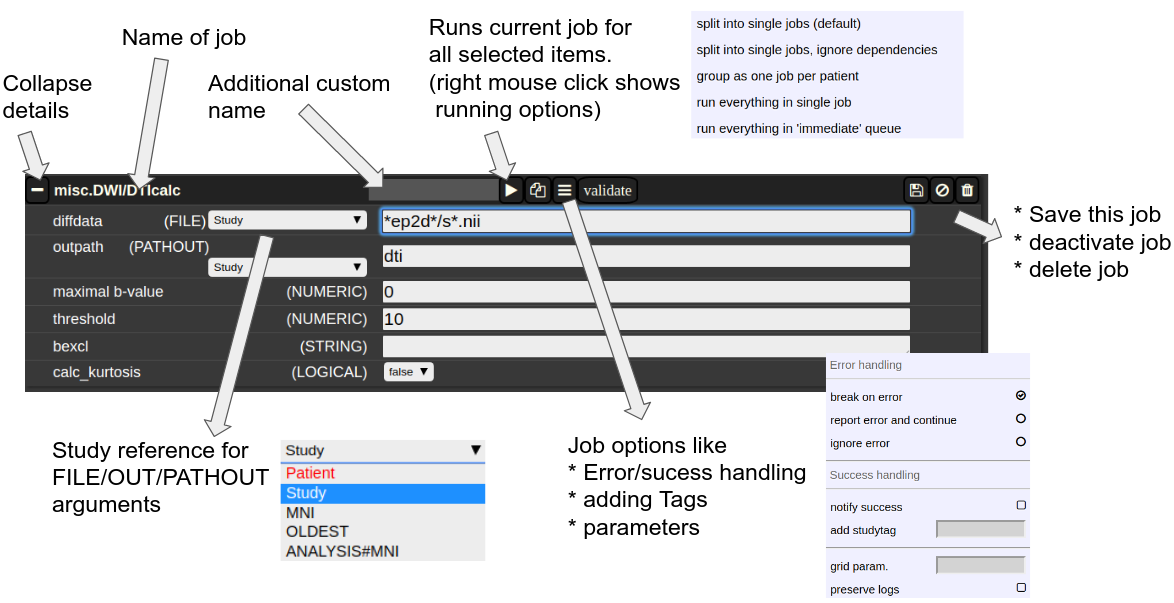
Figure 3: The anatomy of a single job.